هیچ محصولی در سبد خرید نیست.
نمایندگی رسمی Brithish DNW در ایران



















برای **کار توزیع تجهیزات پزشکی در ایران** چند نکته مهم هست:
### 1. آیا برای شرکت توزیعی جواز IMED لازم است؟
بله.
هر شرکتی که بخواهد **تجهیزات پزشکی را توزیع یا عرضه کند** باید در **سامانه IMED اداره کل تجهیزات پزشکی** ثبتنام کند و **مجوز فعالیت توزیعی** بگیرد. بدون ثبت در IMED عملاً امکان فعالیت رسمی و فروش به مراکز درمانی وجود ندارد.
### 2. آیا استاندارد **ISO 13485** لازم است؟
برای **شرکتهای صرفاً توزیعی معمولاً الزامی نیست**.
ISO 13485 بیشتر برای موارد زیر الزامی یا بسیار مهم است:
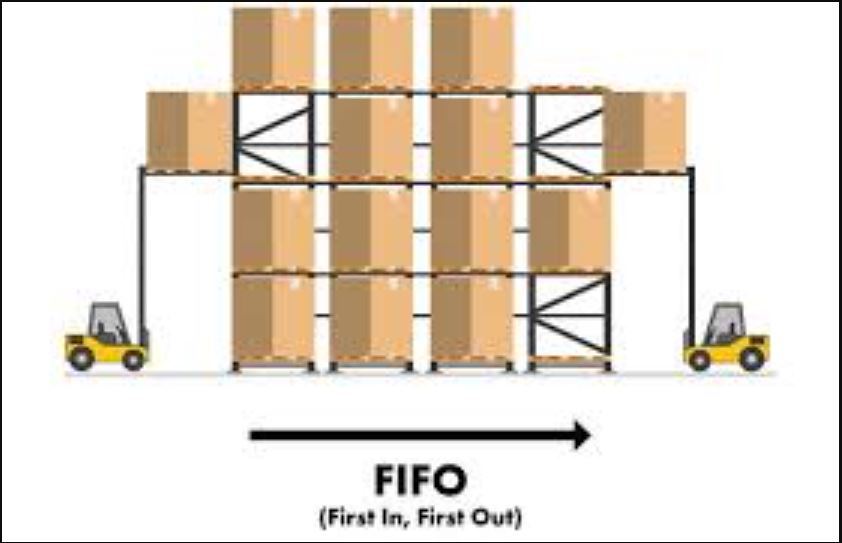
– تولیدکنندگان تجهیزات پزشکی
– شرکتهای طراحی و ساخت
– گاهی برای واردکنندگان یا نمایندگیها (در برخی شرایط)
اما برای **توزیعکننده صرف** معمولاً داشتن آن **اجباری نیست**، هرچند داشتن سیستمهای کیفیت (مثل ISO 9001 یا رویههای GDP) میتواند امتیاز محسوب شود.
### 3. آیا اسکوپ کاری و نوع تجهیزات جداگانه ثبت میشود؟
بله. در IMED:
– **هر محصول یا تجهیز پزشکی باید جداگانه ثبت شود.**
– شرکت توزیعی فقط میتواند **محصولاتی را توزیع کند که در پروفایل آن شرکت در IMED ثبت شدهاند**.
– معمولاً ثبت بر اساس:
– **نام محصول**
– **کلاس خطر (A,B,C,D)**
– **کمپانی سازنده**
– **مدل یا خانواده محصول**
### 4. آیا شرکت توزیعی میتواند همه کلاسهای خطر را بفروشد؟
در اصل **بله**، اما با شرطها:
– محصول باید **در IMED ثبت شده باشد**.
– شرکت باید **نمایندگی یا مجوز توزیع آن محصول را داشته باشد**.
– برای کلاسهای **خطر بالاتر (C و D)** معمولاً:
– مستندات بیشتر
– کنترلهای بیشتر
– گاهی شرایط نگهداری یا خدمات خاص لازم است.
 خلاصه:
خلاصه:
IMED:لازم است
ISO 13485:** برای توزیعکننده معمولاً اجباری نیست
اسکوپ تجهیزات:** هر محصول جداگانه در IMED ثبت میشود
کلاس خطر:** محدودیت مطلق ندارد ولی ثبت و مستندات لازم است
اگر بخواهید، میتوانم هم توضیح بدهم:
-شرایط گرفتن مجوز شرکت توزیعی تجهیزات پزشکی**
مدارک لازم در IMED برای شرکت توزیعی**
-ساختار اسکوپ توزیع در اداره تجهیزات پزشکی**.